Job Submission
HawkDock requires PDB files as the input. Users should provide their own PDB files or enter a PDB ID (Non-standard amino acids, heteroatoms, original hydrogens and residues with incomplete backbone atoms will be deleted. Besides, missing side chains and polar hydrogens will be automatically added.). For HawkDock jobs, the receptor will be stationary in the predictions while the ligand will be moved. So we should submit the larger protein as the receptor.
PDB ID: If you want to use the entire pdb, you can just enter a PDB ID (e.g. 1C3D). Otherwise, you need to specify the chain ID you want to dock (e.g. 1C3D:A, 1V8Z:AB).
Re-rank top10 models by MM/GBSA : MM/GBSA has a better re-ranking capability. However, it takes much longer than the default settings and thus only top 10 models are supported now. In addtion, the key residues of the top 10 models will also be shown. Notice: Since all protein residues will be renumberd from 1 by HawkDock, the top 10 key residues number may be different from the original PDB. Besides, the chain ID of receptor and ligand will be set to A and B by HawkDock, respectively.
Restraints: Users need to provied such information on one line in the text box like
97:A;134:A;10, 98:A;138:A;9
number (receptor):chain ID (receptor);number (ligand):chain ID (ligand);distance
The distance of residue 97 of chain A on the receptor and residue 134 of chain A on the ligand will be within 10 Å. The distance of residue 98 of chain A on the receptor and residue 138 of chain A on the ligand will be within 9 Å. Notice: The residues in a line must be separated by comma.
Job Output
If you have provided a valid email address, an email will be sent to you with a link to the result page when your job is finished.
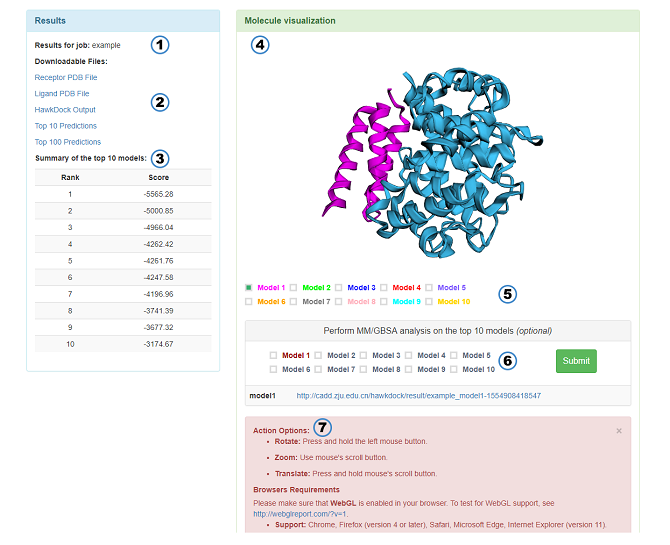
The result page features:
-
Results for job: Name of the job.
-
Downloadable Files: The PDB files used as the docking input, the HawkDock output file (The lower scores, the better predictions. If the low score is behind the high score, it indicates that the result is from cluster), and PDB files of the top 10 and top 100 predictions whose hydrogens are deleted. Notice: All protein residues will be renumberd from 1 by HawkDock. In addition, the chain ID of receptor and ligand will be set to A and B by HawkDock, respectively.
-
Summary of the top 10 models: The score of the top 10 models.
-
Molecule visualization: A 3Dmol.js-enabled visualization window shows the top 10 models.
-
Check box: The option can control 3Dmol.js to choose which models to show.
-
MM/GBSA: You can submit some models to calculate MM/GBSA. Red indicates that the model has been submitted. The URLs of results are shown below.
-
Instructions of 3Dmol.js: A brief introduction of the mouse's usage in the 3Dmol.js and the browsers requirements of 3Dmol.js.
If you have provided a valid email address, an email will be sent to you with a link to the result page when your job is finished.
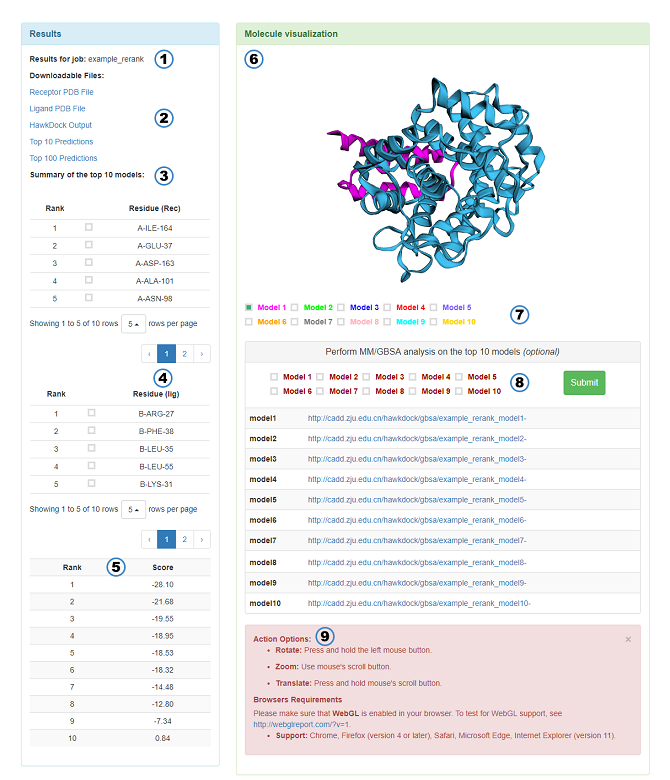
The result page features:
-
Summary of the top 10 models:
- 10 most frequently occurring residues of receptor in the top 10 residues of the top 10 models. The optional options in the table allow users to choose which residue to appear in the 3Dmol.js. Notice: If a reliable model can be founded, the MM/GBSA results of this model are more reliable than the results base on the top 10 models.
- 10 most frequently occurring residues of ligand whose features are similar to the receptor.
- The binding free energy (kcal/mol) calculated by MM/GBSA of the top 10 models.
-
Other features are the same as above.
Job Submission
MM/GBSA is employed to predict the binding free energy and decompose the free energy contributions to the binding free energy of a protein–protein complex in per-residue.
The PDB files or PDB ID are required to be input by users (Non-standard amino acids, heteroatoms, and original hydrogens will be deleted. Besides, missing heavy atoms and new hydrogens will be added by tleap.). The uploaded protein-protein complex structures need to be docked in advance or be determined by experimental techniques. If you model the protein-protein complex structures by HawkDock, you can assign "A" and "B" to the chain ID of receptor and ligand, respectively. If not, you need to specify the chain ID according to the complex you upload.
-
PDB ID: Enter the PDB ID of your complex (e.g. 1SYX).
-
Chain ID: Specify the chain ID according to the uploaded complex (e.g. A). If the receptor and ligand are not a monomer, ";" is requested to separate the chain ID (e.g. A;B or A;B;C). In addition, chain ID must be single character and unique.
-
Parameters: The MM/GBSA is calculated based on the ff02 force field, the implicit solvent model and the GBOBC1 model (interior dielectric constant = 1). The whole system was minimized for 5000 steps with a cutoff distance of 12 Å for van der Waals interactions (2000 cycles of steepest descent and 3000 cycles of conjugate gradient minimizations).
Job Output
If you have provided a valid email address, an email will be sent to you with a link to the result page when your job is finished.
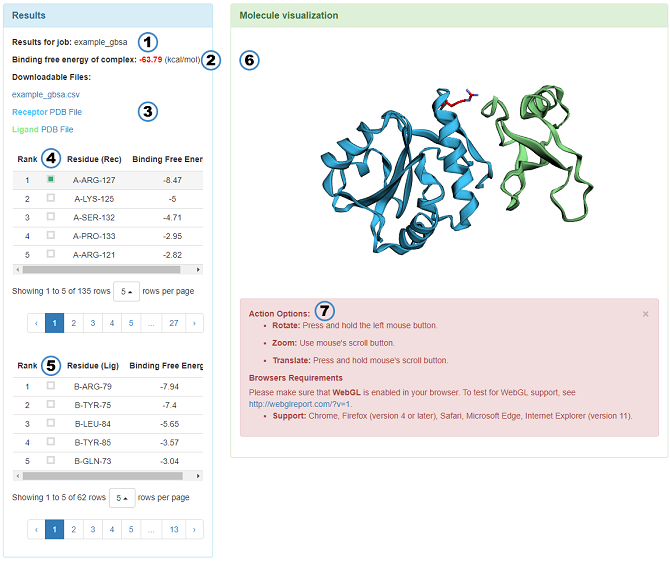
The result page features:
-
Results for job: Name of the job.
-
Binding free energy of complex: The value of binding free energy of protein-protein complex.
-
Downloadable Files: Result of the MM/GBSA, the pre-proccessed PDB files of receptor and ligand. The contents of csv file are as follows.
-
RESIDUE ID: "R" and "L" mean receptor and ligand, respectively.
-
VDW: Van der Waals potentials.
-
ELE: Electrostatic potentials.
-
GB: Polar Solvation free energies predicted by the Generalized Born model.
-
SA: Nonpolar contribution to the solvation free energy calculated by an empirical model.
-
TOTAL: Final estimated binding free energy (kcal/mol) calculated from the terms above. (The lower binding free energy, the more critical residues)
-
-
The upper table: The energy contributions of receptor in per-residue from large to small is shown in the table. The optional options in the table allow users to choose which residue to appear in the 3Dmol.js.
-
The lower table: The table of ligand whose features are similar to the receptor.
-
Molecule visualization: A 3Dmol.js-enabled visualization window shows the complex structure.
-
Instructions of 3Dmol.js: A brief introduction of the mouse's usage in the 3Dmol.js and the browsers requirements of 3Dmol.js.